Статьи
Границы | Повышение точности RNA-Seq с помощью MapAl | генетика
- 1. Введение RNA-Seq использует секвенирование кДНК следующего поколения для изучения экспрессии генов....
- 2.1.1. Комплект 1
- 2.1.2. Набор 2
- 2.2. MapAl Pipeline
- 2,3. Количественная оценка уровней выражения
- 2,4. Меры воспроизводимости
- 3. Результаты
- 3.1. Чтение карт
- 3.2. Воспроизводимость количественного выражения профилирования
- 3.3. Сравнение времени выполнения
- 4. Дискуссия
- Примечание добавлено в доказательство
- Наличие и реализация
- Заявление о конфликте интересов
- Подтверждения
- Рекомендации
- аппендикс
- Детальное сравнение оценок уровня экспрессии по двум конвейерам
1. Введение
RNA-Seq использует секвенирование кДНК следующего поколения для изучения экспрессии генов. Он был применен, чтобы получить глобальные представления о сложности транскриптома ( Cloonan et al., 2008 ; Мортазави и др., 2008 ; Ramsköld и др., 2009 ; Тан и др., 2009 ). В отличие от других технологий профилирования, RNA-Seq может предоставить комплексный анализ экспрессии генов, который не зависит от зондов для мишеней, которые должны быть указаны заранее. Он особенно хорошо подходит для открытия экзонов и сплайс-соединений de novo и позволяет проводить качественное профилирование экспрессии в геноме организмов с неизвестной последовательностью генома.
Все чаще проявляется интерес также к применению RNA-Seq для количественной оценки экспрессии генов ( Удар, 2009 ). В то время как более ранняя работа была сосредоточена на чтениях, которые однозначно идентифицируют стенограмму ( Вильгельм и др., 2008 ), улучшенные алгоритмы позволяют расширить анализ данных на сложные генные модели альтернативного сплайсинга, также принимая во внимание большое количество считываний, которые могут происходить из разных форм сплайсинга ( Цзян и Вонг, 2009 ). Различение сложных альтернативных форм сплайсинга теперь возможно с помощью современных инструментов, таких как ERANGE ( Мортазави и др., 2008 ), ALEXA-seq ( Гриффит и др., 2010 ) НЕУМА ( Ли и др., 2011 ), IsoEM ( Николае и др., 2010 ), РСЭМ ( Ли и др., 2010 ; Ли и Дьюи, 2011 ) или TopHat + Запонки ( Трапнелл и др., 2009 , 2010 ).
В популярном наборе инструментов TopHat + Cufflinks это достигается путем предварительного выравнивания операций чтения с геномом, с открытием de novo экзонов и соединений сплайсинга ( TopHat ). Затем эта информация используется для сбора транскриптов и расчета их численности на втором этапе ( запонки ). Запонки могут использовать преимущества аннотации эталонной генной модели для количественного определения известных транскриптов, пропуская этап сборки транскрипта.
Инструмент MapAl , представленный здесь, основан на этом двухэтапном подходе и расширяет алгоритм, чтобы уже использовать генные модели на этапе выравнивания. Отсутствие необходимости идентифицировать новые формы сплайсинга для известных генов значительно улучшает назначение операций чтения аннотированным транскриптам, особенно для форм сплайсинга, охватываемых только небольшим числом операций чтения ( Łabaj et al., 2011 ). Этот новый подход значительно увеличивает количество транскриптов, которые могут быть надежно измерены. Это представляет общий интерес, поскольку точность измерения определяет мощность любого последующего анализа, такого как чувствительное обнаружение дифференциально выраженных транскриптов, независимо от того, используются ли реплики или нет ( Андерс и Хубер, 2010 ).
2. Материалы и методы
2.1. Источники данных и аннотации
Чтобы проверить предложенный подход, мы рассмотрим два набора данных, представляющих разные технологии, с различной длиной считывания, глубиной считывания, стратегиями секвенирования (односторонним и парным концом), типом репликации и полученными из разных линий клеток человека.
2.1.1. Комплект 1
Было выполнено три повторных измерения мРНК, выделенной из культуры линии клеток HMEC 184A1 человека. С общим числом операций чтения в 993 млн. 50 б.п., что соответствует целой проточной ячейке ABI SOLiD-3 + на образец измерения, это представляет собой один из самых больших наборов данных RNA-Seq с техническими дубликатами на сегодняшний день (измерения SRR413934, SRR413935 и SRR413936 от проект PNNL-EMSL с SRA-ID SRP011007).
2.1.2. Набор 2
Были также исследованы три измерения из общедоступных в настоящее время профилей экспрессии клеточных линий H1-hESC из лаборатории Wold / Caltech (эксперименты SRX026674, SRX026669 и SRX026685 из проекта ENCODE с SRA-ID SRP003497). Они обеспечивают 2 × 50 миллионов парных чтений на уровне 75 п.н. за измерение, полученных с помощью анализатора Illumina Genome Analyzer II. Хотя измерения не являются техническими копиями, биологические копии клеточных линий достаточно похожи (в отличие, скажем, от образцов пациентов), чтобы можно было сравнительно изучить методы обработки и их влияние на точность измерений. Тот факт, что мы наблюдаем одинаково сильное улучшение производительности в обоих наборах данных, также подтверждает этот случай .
Для беспристрастной оценки скорости идентификации формы сплайсинга мы сосредоточились на чтениях, приведенных в соответствие со всеобъемлющими 140 079 человеческими транскриптами, аннотированными в EnsEMBL 58.
2.2. MapAl Pipeline
Производительность MapAl демонстрируется с использованием хорошо известных программ TopHat + Cufflinks в качестве эталона. Аналогичные результаты наблюдались с альтернативными программами (данные не показаны).
В стандартном конвейере TopHat + Cufflinks аннотированные «генные модели» используются только на этапе Cufflinks (правая часть, рисунок 1 А). MapAl , напротив, позволяет эксплуатировать генные модели уже на этапе выравнивания: чтения непосредственно выровнены с известными последовательностями транскриптов (левая сторона, рисунок 1 Б). Затем MapAl сопоставляет выровненные чтения с геномными местоположениями, описанными соответствующими «моделями генов» (выделено полужирным шрифтом), беря в качестве входных данных файл сопоставления транскриптома (SAM) вместе с аннотациями транскрипта, и создает файл SAM на основе хромосомы. Запонки могут впоследствии использоваться для оценки содержания стенограммы (правая часть).
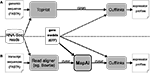
Рисунок 1. Рабочие процессы для профилирования выражений RNA-Seq . Установленный конвейер TopHat (A) сравнивается с новым подходом MapAl (B) , который использует генные модели уже на этапе выравнивания. Стрелки указывают поток данных - выравнивания могут быть предоставлены в формате SAM (выравнивание последовательностей / карта), модели генов в GTF (формат передачи генов), уточнение общего формата признаков (GFF).
Чтения, отображающиеся на разные сплайс-формы одного и того же гена, сводятся к одному выравниванию во время обработки MapAl, когда они совпадают с одним и тем же местоположением генома (рисунок 2 ). С другой стороны, различные операции чтения, сопоставленные с несколькими целями, сохраняются как отдельные, соответствующие различным геномным местоположениям.
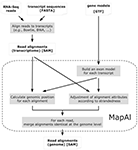
Рисунок 2. Структура алгоритма MapAl . Блок-схема представляет алгоритмическую структуру конвейера MapAl . Шаги, выполняемые сценарием MapAl , отмечены пунктирным прямоугольником с закругленными углами. В частности, чтения сначала выровнены по известным последовательностям транскрипта с использованием любого выравнивателя, поддерживающего формат вывода SAM. Затем они отображаются в геномные местоположения с помощью MapAl с помощью предоставленных моделей генов. Атрибуты выравнивания, встроенные в формат SAM, корректируются в соответствии с многопоточностью. Затем для каждого считывания выравнивания, идентичные на уровне генома, объединяются для получения окончательного файла выравнивания генома.
Мы делаем так, чтобы можно было использовать любой выравниватель чтения, поддерживающий формат SAM ( Ли и Гомер, 2010 ). Здесь, чтения РНК-Seq были выровнены с последовательностями транскрипта с Bowtie ( Лангмид и др., 2009 ). Это облегчает прямое сравнение протестированных конвейеров, потому что Bowtie также используется TopHat для внутреннего использования. Bowtie v0.12.7 и TopHat v1.1.4 были запущены с настройками, подходящими для исследуемых типов наборов данных (подробности см. В приложении).
2,3. Количественная оценка уровней выражения
Уровни экспрессии рассчитывали, используя Запонки v0.9.1 ( Трапнелл и др., 2010 ) с моделями генов EnsEMBL, предоставленными как указано. Для обнаружения транскриптов de novo генные модели EnsEMBL были предоставлены запонкам, чтобы игнорировать все прочтения, которые могли быть получены из известных генов. Параметры были установлены для максимальной чувствительности (–min-frags-per-transfrag 1 и -F 0). При обработке выравниваний в конвейере MapAl также можно установить параметр -A 0, поскольку известно, что все части чтения происходят из одной и той же последовательности транскрипта (этот параметр обычно используется для поддержки надежного обнаружения стыкового соединения через TopHat ). Для форм сращивания, поддерживаемых менее чем одним выравниванием чтения, назначенным запонками , уровни выражений были установлены на ноль. Для прямого сравнения уровня формы сплайсинга результатов MapAl и TopHat оценки выражений должны были быть нормализованы по общему количеству выравниваний, рассмотренных на соответствующем этапе запонок .
2,4. Меры воспроизводимости
Для систематической оценки воспроизводимости мы можем рассмотреть коэффициент вариации (CV) в линейной шкале или SD уровней логарифмической экспрессии. По ряду причин данные по экспрессии генов обычно анализируются в логарифмическом масштабе, на котором различия в экспрессии проверяются с помощью t- теста. Различия в логарифмическом масштабе затем соответствуют кратному изменению в линейном масштабе. В этом контексте подходящим показателем точности является SD в логарифмическом масштабе. Когда мы ссылаемся на относительную ошибку в рукописи, равную 20% или менее, мы пороговую величину SD σ <log2 (120%), чтобы значение μ + σ по сравнению с μ в шкале log2 соответствовало относительной ошибке 20% или меньше по линейной шкале. Мы считали расшифровку стенограммы достоверной, если относительная ошибка составляла менее 20%. Сравнения воспроизводимости не зависят от этого произвольного порога. Обратите внимание, что воспроизводимость также определяет силу статистики, которая работает без повторов ( Андерс и Хубер, 2010 ).
Следует отметить, что многие анализы, учитывающие точность повторения, исключают измерения без сигнала ни в одном из повторов. Это создает смещение методов в сторону лучшей воспринимаемой точности в анализе. В исследуемом наборе данных 1 из всех идентифицированных целей транскриптов 14% имели нулевое чтение в одном или двух повторностях, но ненулевое число в других, при этом наблюдалось 1–26 чтений. Эти расшифровки в значительной степени способствуют измерению шума при низких уровнях экспрессии и, следовательно, должны учитываться как доля ненадежных измерений. Этот подход является последовательным, потому что они имеют бесконечную ошибку в логарифмическом масштабе (а также коэффициент вариации в линейном масштабе всегда> 80%).
3. Результаты
Мы представляем MapAl , новый подход для количественного определения количества транскриптов по данным RNA-Seq. Мы проверили наш инструмент по сравнению с популярным конвейером анализа TopHat + Cufflinks . В частности, мы рассмотрели количество транскриптов, которые можно было бы идентифицировать и надежно измерить. Это было проверено на двух независимых наборах данных, представляющих альтернативные стратегии секвенирования и технологически разные платформы.
3.1. Чтение карт
Считать статистику выравнивания приведены в таблице 1 , С одной стороны, установленный конвейер с TopHat давал до 10% больше отображенных чтений. Это отражает дополнительные, неизвестные транскрипты, обнаруженные путем выравнивания с последовательностью генома. Эффект был особенно очевиден для набора данных 2, где можно было использовать парные операции чтения, и TopHat мог использовать преимущества более длинных операций чтения в 75 бит / с, для которых он был разработан. В отличие от этого, однако, MapAl идентифицировал в 2-3 раза больше чтений, приходящихся на известные соединения экзонов, что является следствием использования известных последовательностей сплайс-формы уже на стадии выравнивания. Выравнивания в типичном транскрипте иллюстрируют этот момент на рисунке A1 в приложении. В окне браузера IGV ( Робинсон и др., 2011 ), синие прямоугольники представляют второй и третий экзоны ENST00000377403 (H6PD). Полоса покрытия в верхней части указывает, что чтения, полностью попадающие в экзоны, отображаются одинаково как MapAl (вверху), так и TopHat (внизу). Разница заключается в выравнивании операций чтения, соединяющих сплайсинговые соединения. В частности, для этой экзоновой структуры никакие чтения, охватывающие экзоны один и два, не были идентифицированы TopHat , тогда как существенное число было правильно отображено MapAl (мост соединения слева от окна браузера). Точно так же TopHat не идентифицировал чтения, охватывающие три и четыре экзона, тогда как MapAl правильно идентифицирует многие чтения, охватывающие эту область (соединяя переход справа от окна браузера). В то время как некоторые операции чтения, охватывающие соединение сплайсинга между двумя и тремя экзонами, были обнаружены TopHat , примерно в два раза больше таких операций чтения было выявлено конвейером MapAl . Именно эти операции считывания в соединениях экзонов часто играют ключевую роль в идентификации выражения конкретной формы соединения, и, следовательно, определяют как скорость идентификации формы соединения, так и точность измерения, специфичную для транскрипта.
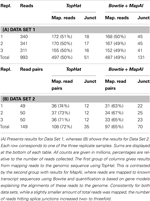
Таблица 1 Статистика чтения и отображения результатов .
Кроме того, мы рассмотрели эффекты предоставления известных соединений соединения TopHat через файл GTF (опция -G) или через файл списка соединений (опция -j). Интересно, что это последовательно увеличивает количество выравниваний, приходящихся на стыковые соединения, примерно на 7%. Таким образом, оставшееся несоответствие может быть объяснено только особенностями алгоритма TopHat , такими как фильтрация операций чтения, когда соединение попадает в конечную область операций чтения. Мы также исследовали альтернативные варианты параметров TopHat (такие как менее строгие настройки -a и -F), но основная картинка остается, давая только дальнейшее увеличение на 8% числа операций чтения, приходящихся на соединения.
Также стоит отметить, что для MapAl около 80% идентифицированных стыковых соединений были поддержаны более чем 10 выравниваниями. Для TopHat эта доля составляла около 2/3, что указывает на то, что MapAl не только может идентифицировать больше операций чтения, приходящихся на соединения сплайсинга, но также увеличивает поддержку идентифицированных соединений сплайсинга.
3.2. Воспроизводимость количественного выражения профилирования
Таблица 2 сравнивает количество транскриптов, которые могут быть идентифицированы и надежно измерены. Для набора 1 конвейер MapAl обнаружил 101 169 сплайс-форм против 87 649, идентифицированных TopHat (72 против 63% всех известных транскриптов). См. Приложение для подробного сравнения.

Таблица 2 Статистика выявленных и достоверно измеренных транскриптов .
Еще более явные различия в производительности возникают, когда мы рассматриваем только надежно измеренные формы сплайсинга: стандартный конвейер TopHat + Cufflinks , включая обнаружение de novo альтернативных форм сплайсинга и генов, может оценить 35 405 форм сплайсинга с относительной погрешностью <20%. Интересно, что использование известных моделей генов увеличило количество надежно измеренных транскриптов до 39,116, хотя теперь они включают только известные гены и формы сплайсинга.
Однако с помощью MapAl уровни экспрессии 56 980 транскриптов можно было бы надежно измерить, получив улучшение почти на 50% по сравнению с установленным рабочим процессом. фигура 3 сравнивает распределения ошибок измерений. С одной стороны, максимальные ошибки больше для TopHat (красные кривые, идущие дальше вправо). С другой стороны, MapAl может измерить большее количество транскриптов с низкими ошибками (черные кривые показывают более высокие значения на осях Y слева от пунктирных линий). Пунктирные линии показывают ошибки измерения 20%. Эти наблюдения в равной степени применимы к техническим копиям набора данных 1 и биологическим копиям набора данных 2 (которые в целом демонстрируют более высокие вариации).
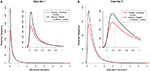
Рисунок 3. Распределение ошибок измерений . На каждом из графиков сравниваются распределения ошибок измерений для TopHat (красная линия) и MapAl (черная линия). Ось X показывает SD по трем повторным измерениям в логарифмическом масштабе. Пунктирная вертикальная линия отмечает ошибку в 20%. Ось Y представляет количество форм сплайсинга, которые можно измерить с определенной точностью (масштабируется до произвольной единицы, позволяющей сравнивать выборки). Первая панель отображает результаты для набора данных 1 [ (A) технические копии], вторая панель для набора данных 2 [ (B) биологические копии].
Следует отметить, что 56 980 известных транскриптов, которые можно надежно измерить, можно легко дополнить измерениями для вновь обнаруженных генов, добавив еще 11 288 транскриптов и доведя общее количество до 68 268 надежно профилированных форм сплайсинга. Это почти вдвое больше, чем 35,405, которые можно было бы надежно оценить с помощью стандартного конвейера, и аналогичные улучшения могут быть продемонстрированы для набора данных 2 (таблица 3 ).

Таблица 3 Статистика идентифицированных и достоверно измеренных транскриптов, добавление генов, идентифицированных de novo .
Поскольку всегда интересно сравнивать производительность альтернативных конвейеров, MapAl был сконструирован для простого сочетания с другими инструментами, поддерживающими формат SAM, поддерживающими будущие разработки и дальнейшие независимые тесты.
3.3. Сравнение времени выполнения
Собранные наборы данных становятся еще больше, интересно оценить время выполнения анализируемых конвейеров. В рассмотренных подходах учитывалось одинаковое количество выравниваний, что давало одинаковое время выполнения этапа Запонки . Поскольку этот шаг является быстрым по сравнению с общим временем работы конвейера, мы можем сосредоточиться на остальных шагах, сравнивая Bowtie + MapAl и TopHat .
Поскольку производительность MapAl была привязана к диску, распараллеливание не дало значительного ускорения. Таким образом, MapAl был запущен в однопоточном режиме, однако параллельный анализ операций чтения, выровненных по транскриптам для каждой хромосомы, заслуживает дальнейшего рассмотрения. Обратите внимание, что производительность файловой системы может легко стать ограничивающим фактором в общей пропускной способности.
Таблица 4 представляет усредненное время выполнения для обоих конвейеров. Для набора данных 1 с более коротким односторонним чтением конвейер MapAl выполнялся в пять раз быстрее, чем установленные запонки TopHat +. Для набора данных 2 MapAl был почти в два раза быстрее, несмотря на два фактора, работающих на преимущество установленных инструментов: (1) TopHat был спроектирован и оптимизирован для более длинных чтений 75 б.п., и (2) каждая пара чтений рассматривается как TopHat «один фрагмент», в то время как оба чтения обрабатываются независимо MapAl , удваивая эффективное число последовательностей, которые необходимо учитывать.

Таблица 4 Сравнение времени выполнения трубопровода .
4. Дискуссия
В то время как использование преимуществ известных последовательностей транскриптов на стадии выравнивания и одновременное обнаружение новых альтернативных форм сплайсинга известных генов потребует разработки расширенных моделей для оценки уровней транскриптов, MapAl опирается на существующие инструменты, чтобы обеспечить быстрое и простое решение для количественного выражения. профилирование по RNA-Seq.
Он поддерживает как пользователей, так и дальнейшее развитие, предоставляя свободный выбор сочетания альтернативных этапов на разных этапах процесса. В частности, можно использовать широкий диапазон карт чтения, поддерживающих стандартный формат SAM, потому что MapAl также правильно обрабатывает Indels. Инделс - самая частая форма ошибки секвенирования ( Альберс и др., 2011 ) но может также играть важную роль в открытии вариантов ( Кравитц и др., 2010 ).
Для этой рукописи программное обеспечение было проверено на данных из односторонних операций чтения, сгенерированных с помощью многопоточного протокола RNA-Seq (SOLiD, набор данных 1), и на данных от парных операций чтения, сгенерированных с помощью нецепного протокола RNA-Seq (Illumina). , Набор данных 2). В настоящее время мы расширяем инструмент для поддержки одностороннего, парного и парного считываний как из многопоточных, так и из нецепочечных протоколов RNA-Seq.
Мы также улучшаем обработку экзоновых переходов. За редким исключением, экзоны длиннее 50 нуклеотидов ( Бергет, 1995 ). Таким образом, чтения, охватывающие более двух экзонов, были очень редкими для ранних данных секвенирования следующего поколения, с типичной длиной считывания 36 бп. Текущее оборудование уже производит чтение 150 б.п. С увеличением продолжительности чтения на современных платформах чтение, охватывающее несколько соединений сплайсов, становится все более частой проблемой. Поскольку эти операции чтения особенно эффективны при распознавании определенных форм соединения, мы добавляем полную поддержку операций чтения, охватывающих несколько соединений.
Следующая версия нашего программного обеспечения, реализующего эти функции, станет доступна на http://www.bioinf.boku.ac.at/pub/MapAl/early Март 2012 года, а также будет поддерживать последнюю версию запонок (январь 2012 года), используя преимущества последних функций, таких как улучшенная коррекция многократного чтения, которая была представлена в прошлом году.
Таким образом, MapAl предоставляет гибкий модульный подход к количественному профилированию выражений RNA-Seq, опираясь на сильные стороны популярных устоявшихся инструментов. Он реализует расширенные функции, связанные с проблемами выравнивания чтения, для удовлетворения потребностей анализа последних платформ секвенирования. В частности, он использует информацию о последовательности в форме соединения уже на этапе выравнивания. MapAl увеличивает количество достоверно измеренных известных транскриптов примерно на 50%, а также позволяет проводить профилирование новых генов, что почти вдвое увеличивает количество транскриптов, которые можно надежно оценить.
Примечание добавлено в доказательство
С версией 1.4.0, выпущенной в этом году, TopHat теперь может также отображать чтения непосредственно в транскриптом, используя идеи, аналогичные подходу, впервые введенному в Łabaj et al. (2011) , Начальные сравнения с MapAl , однако, предполагают значительные различия в итоговых оценках уровня экспрессии, по-видимому, из-за дополнительной эвристики и различных деталей реализации. Ввиду существенного влияния выбора одной программы на другую, дальнейшие сравнительные исследования альтернативных инструментов, безусловно, представляют интерес. Модульный подход MapAl предоставляет пользователям ценный выбор, позволяя легко комбинировать эффективную двухэтапную стратегию сопоставления с установленными параметрами, такими как Bowtie и Cufflinks , а также напрямую работать с другими инструментами, поддерживающими формат SAM.
Наличие и реализация
Пакет MapAl доступен для загрузки под лицензией GPL по адресу www.bioinf.boku.ac.at/pub/MapAl
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось в отсутствие каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Подтверждения
Авторы выражают благодарность доктору Лан Ху (Центр вычислительной биологии рака, Институт рака Дана-Фарбер, Бостон) за ее вклад в улучшение MapAl и ее помощь в тщательном тестировании инструмента. Эта работа была поддержана Венским научно-техническим фондом (WWTF), Baxter AG, Австрийским технологическим институтом и Австрийским центром биофармацевтических технологий. Часть этого исследования была выполнена с использованием EMSL, национального научного учреждения-пользователя, спонсируемого Управлением биологических и экологических исследований Министерства энергетики США, расположенного в Тихоокеанской северо-западной национальной лаборатории. Bryan E. Linggi был частично поддержан финансированием LDRD от Тихоокеанской Северо-Западной Национальной Лаборатории.
Рекомендации
>
аппендикс
Параметры выполнения Bowtie и TopHat
Чтобы облегчить прямое сравнение установленного конвейера TopHat с новым подходом, реализованным в MapAl, мы использовали Bowtie в качестве выравнивателя, потому что он также используется TopHat . Чтобы удостовериться, что сравнение максимально справедливо, мы запустили Bowtie с настройками, которые были использованы внутренне протестированным TopHat (v1.1.4).
Для набора данных 2 (Illumina) TopHat запускает Bowtie со следующими параметрами:
bowtie -q –un TopHat1 / tmp / left_kept_reads_
отсутствующий.
–Max / dev / null -n 2 -p 15 -k 40 -m 40
Поскольку мы не заинтересованы в отдельных отчетах о несогласованных чтениях и чтениях, выравнивающих слишком много местоположений, мы указываем опции –max и –un, чтобы отбросить их. Ключ -S запрашивает вывод выравниваний в формате SAM:
bowtie -q -n 2 -p 15 -k 40 -m 40 -S
Для набора данных 1 (ABI SOLiD, colourspace) TopHat запускает Bowtie со следующими параметрами:
bowtie -q -C –col-keepends –un \
TopHat1 / TMP / left_kept_reads_missing.fq
-Максимум \
/ dev / null -n 2 -p 15 -k 40 -m 40
Поскольку исходные файлы этого набора данных предоставляются в. Фастк и. Качественные форматы, мы меняем -q на -f. Опять же, мы опускаем –max и –un и добавляем -S, чтобы выбрать необходимые выходные файлы и форматы, получая:
bowtie -f -C –col-keepends -n 2 -p 15 -k
40-м 40-с
Пользователям может потребоваться увеличить значения параметров -k и -m при одновременном выравнивании непосредственно с транскриптомом, чтобы можно было сообщать о более правильных попаданиях, поскольку для сложных структур в форме соединения можно ожидать большое количество таких совпадений.
Детальное сравнение оценок уровня экспрессии по двум конвейерам
MapAl определяет дополнительные операции чтения, охватывающие экзонные соединения. Здесь мы обсуждаем различия в оценках уровня выражения, которые наблюдаются в результате. Сравнительные графики рассеяния для отдельных копий представлены на рисунке. A2 , Для каждого транскрипта x- оси показывают уровень выражения MapAl по сравнению с уровнем выражения TopHat на y- осях, каждый в масштабе log10. Более темные уровни серого указывают на большее количество транскриптов. Обратите внимание, что мы отображаем стенограммы без вызова выражения в –5, чтобы также визуализировать различия в вызовах присутствия.
Сначала рассмотрим простые генные модели рисунка A3 О. При достаточном покрытии возможно оценить выражение обеих форм соединения даже без чтения, охватывающего экзонное соединение. Следовательно, добавление выравниваний чтения, которые попадают на стыки сплайсинга, немного увеличит покрытие на границах экзонов и, таким образом, увеличит соответствующие уровни экспрессии. Это способствует наблюдению на графиках рассеяния (рис. A2 ), что уровни экспрессии для MapAl в целом выше (плотности ниже диагонали).
Большие различия могут уже ожидаться для простой модели гена рисунка A3 Б. Рассмотрим сценарий, в котором TopHat не может идентифицировать чтения, охватывающие экзон 2, как показано на графике гипотетического покрытия в верхней части панели. Это может произойти особенно легко для коротких экзонов. В этом случае, как представляется, данные свидетельствуют о том, что сплайс-форма T1 была четко выражена, в то время как отсутствуют конкретные доказательства для экспрессии T2. Добавление выравниваний чтения, которые покрывают стыковые соединения между экзонами 1 и 2 или экзонами 2 и 3, изменяет картину. Можно даже прийти к противоположному заключению, а именно, что форма сплайсинга T2 была выражена, тогда как T1 не был, если имеется достаточное количество этих прочтений. В таких крайних случаях будет наблюдаться разница в вызовах присутствия (таблица A1 ). Они вносят вклад в плотности, параллельные оси на графиках рассеяния.
Как правило, если обе формы соединения были выражены, можно ожидать дополнительных выравниваний чтения, которые покрывают соединения сплайсинга между экзонами 1 и 3, предоставляя конкретные доказательства для выражения формы сплайсинга T1, а также дополнительные выравнивания чтения, которые охватывают соединения соединения между экзонами 1 и 2 или экзоны 2 и 3, обеспечивающие конкретные доказательства экспрессии сплайс-формы T2. В этом случае уровень выражения MapAl для T1 будет ниже (что способствует плотности выше диагонали). В отличие от TopHat , MapAl может вызвать вызов присутствия для T2, способствуя горизонтальной плотности на диаграмме рассеяния. Это объясняет, почему плотность, указывающая вызовы присутствия, уникальные для MapAl (горизонтальный), имеет больший объем, чем плотность, указывающая вызовы присутствия, уникальные для TopHat (вертикальный). Диаграммы рассеяния, таким образом, отражают, что MapAl последовательно идентифицирует больше транскриптов. Таблица A1 дает подробное сравнение.
Более сложные эффекты можно понять, используя модель гена на рисунке A3 С в качестве примера. Рассмотрим сценарий, в котором TopHat не может определить никакие чтения, охватывающие экзонные соединения, как показано на графике гипотетического покрытия в верхней части панели. При отсутствии доказательств конкретного выражения различных форм сращивания операции чтения распределяются равномерно. Дополнительные свидетельства от выравнивания, падающего на соединения соединения, играют критическую роль в оценке определенных уровней выражения формы соединения. В общем случае операции чтения, охватывающие соединения экзонов, поддерживают доминирующее выражение конкретной сплайс-формы. Поэтому, принимая во внимание это свидетельство, вы повысите оценку уровня выражений MapAl для этой формы сплайсинга, одновременно снизив оценки уровня выражений для остальных. Это изменение меньше, поскольку оно распространяется по нескольким формам сращивания. Это объясняет, почему отклонения ниже диагонали на диаграммах рассеяния, сравнивающих MapAl и TopHat , сильнее. Фактически, в зависимости от набора данных и рассматриваемого дубликата , уровни экспрессии для MapAl были выше для 38% -42% всех известных транскриптов и выше для TopHat на 13% -17% (таблица A2 ).
Таким образом, правильное рассмотрение операций чтения, охватывающих соединительные формы в MapAl, может значительно повлиять на оценки выражений.
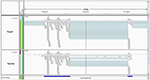
Рисунок А1. Выравнивания в типичной расшифровке . На рисунке показано окно браузера IGV ( Робинсон и др., 2011 ). Синие прямоугольники представляют второй и третий экзоны ENST00000377403 (H6PD). Серая полоса покрытия в верхней части отражает то, что чтения, полностью попадающие в экзоны, отображаются одинаково как MapAl (вверху), так и TopHat (внизу). Разница заключается в выравнивании операций чтения, расположенных на стыке сплайсинга.
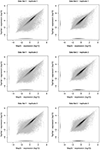
Рисунок А2. Точечный график уровней экспрессии транскрипта . Диаграммы разброса представляют связь между выражением транскрипта, оцененным запонками для конвейера TopHat ( ось y ) и конвейера MapAl ( ось x ). В левом столбце представлены диаграммы рассеяния трех копий набора данных 1, в правом столбце представлены диаграммы рассеяния трех копий набора данных 2. Из всех диаграмм рассеяния ясно, что значительное количество транскриптов выражено выше для конвейера MapAl, чем для TopHat one. ,
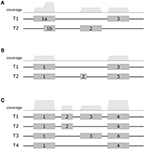
Рисунок А3. Примерные генные модели . Схематические диаграммы представляют три примера генных моделей. В верхнем ряду отображается гипотетическое покрытие, предполагающее равномерное распределение чтений, попадающих исключительно в экзоны. Модели (A – C) демонстрируют возрастающую сложность. В первой модели все еще возможно оценить выражение альтернативных форм сплайсинга даже без показаний, охватывающих соединения экзонов. Дополнительные чтения связующего соединения будут умеренно влиять на оценки уровня выражения. В следующей модели добавление выравниваний чтения, которые попадают на стыки соединений, уже может значительно повлиять на вызовы присутствия и оценки конкретных уровней выражений соединений. В самой сложной модели добавление выравниваний чтения, которые попадают на соединения сплайсинга, играет критическую роль в оценке конкретных уровней выражения сплайс-формы. В этом сценарии дополнительные свидетельства повысят оценку уровня экспрессии для доминантной сплайс-формы, одновременно снизив оценки уровня экспрессии для остальных.

Таблица А1 . Сравнение присутствия звонков .

Таблица А2 . Сравнение оценок уровня экспрессии .
Похожие
Алкоголь: повышение цен может снизить вред и способствовать сбору доходов.Монган, Дейрдре (2013) Алкоголь: повышение цен может уменьшить вред и способствовать сбору доходов. Drugnet Ирландия, выпуск 44, зима 2012, с. 7-9. Алкоголь чувствителен к цене - увеличение стоимости алкоголя сокращает его потребление, а снижение стоимости алкоголя увеличивает его потребление. Поэтому цена часто используется в качестве политического рычага для снижения потребления алкоголя и связанного Официальный Huawei Y5 Prime 2018, скромный смартфон начального уровня
Новый смартфон только что незаметно появился на официальном сайте Huawei: Y5 Prime 2018. Это устройство начального уровня с распознаванием лиц, экраном 18: 9 и хорошим временем автономной работы. конструктор Робко объявлено на АМЕРИГАЗ будет доставлять газовые баллоны с помощью беспилотника
Если вы живете в доме, где используются газовые баллоны - и, согласно данным GUS, это касается 1/3 всех поляков, как и полагается технологической службе, мы представляем вам настоящую технологическую революцию. Есть очень хороший шанс, что ваша жизнь скоро изменится к лучшему. Я никогда не говорил вам об этом, потому что не было никакой возможности, но во время колледжа я попал в компанию людей, увлекающихся курением ... водопровода. Нормально, с Тестирование и сравнение тостеров »10 лучших в мае 2019 года
Согласно тостерному тесту, согласно опыту и отзывам многих клиентов, тостер является одним из самых важных инструментов для завтрака. Тостер предназначен для поджаривания хлеба , что означает, что хлеб поджарен. Тостер может иметь Тест Sony Xperia Z1: наш полный обзор
введение На выставке IFA Sony представила Xperia Z1. С его кодовым названием «Хонами» мы знали о нем все еще до его объявления. Действительно, блогеры и журналисты утопили нас в утечках, к расстройству желудка. К счастью для нас, передозировка заканчивается: Samsung анонсировала Galaxy Note III, Apple выпустила iPhone, а Sony официально представила Xperia Z1. Краткая история звуковых карт. Устройства, без которых компьютеры молчали
Сегодня мы рассматриваем воспроизведение звука на ПК как очевидное. Между тем, только через два года мой первый компьютер мог говорить со мной. Кроме того, его звуковые возможности были очень ограничены. Вы знаете, что такое звуковая карта? Вероятно, еще это оборудование запомнили или вы догадались, по какому месту назначения. Сегодня звуковые карты встроены в материнскую плату каждого компьютера. Однажды, без такой карты, Загрузка и обновление драйверов USB | USB Driver Fix и обновления | Drivers.com
Загрузка и обновление драйверов USB | USB Driver Fix и обновления | Drivers.com Обновления драйвера USB Нужна загрузка драйверов USB для Windows 10, Windows 8, Windows 7, Vista и XP ? Если у вас возникли проблемы с неработающим USB , прочитайте статью ниже, чтобы исправить проблемы с USB. Проблемы с USB часто, но не всегда, связаны с проблемами с драйверами.
Комментарии
Какие результаты дал мой личный тест?Какие результаты дал мой личный тест? Pampers Premium Care в размере 4 (весовой диапазон 7-18 кг) Уход за подгузниками стоимость продукта (около 60 злотых) не соответствует его Разве это не здорово, что у вас может быть аккаунт в Германии, которым можно управлять с помощью приложения на вашем смартфоне или в режиме электронного банкинга?
Разве это не здорово, что у вас может быть аккаунт в Германии, которым можно управлять с помощью приложения на вашем смартфоне или в режиме электронного банкинга? Кроме того, карта MasterCard, которая дает вам дополнительные возможности при электронных транзакциях и инкассации? Небольшое примечание - Также в других европейских странах есть люди, которые точно хотят создать учетную запись в Германии. Почему нет? Профилактика лучше лечения, а мы в Германии знаем лучше. Качество звука само собой разумеющееся, но как насчет времени автономной работы, скорости зарядки, поддержки подключения или цены?
Качество звука само собой разумеющееся, но как насчет времени автономной работы, скорости зарядки, поддержки подключения или цены? Здесь у нас есть краткое руководство о том, что следует учитывать при покупке портативного динамика, а также наши 13 текущих любимых. Качество звука Первое, на что нужно обратить внимание, это очевидное качество звука, но его сложно измерить, не в последнюю очередь потому, что некоторые производители заглушают вас дикими претензиями и даже
Вы знаете, что такое звуковая карта?
Com Обновления драйвера USB Нужна загрузка драйверов USB для Windows 10, Windows 8, Windows 7, Vista и XP ?
Какие результаты дал мой личный тест?
Разве это не здорово, что у вас может быть аккаунт в Германии, которым можно управлять с помощью приложения на вашем смартфоне или в режиме электронного банкинга?
Кроме того, карта MasterCard, которая дает вам дополнительные возможности при электронных транзакциях и инкассации?
Почему нет?
Качество звука само собой разумеющееся, но как насчет времени автономной работы, скорости зарядки, поддержки подключения или цены?
Качество звука само собой разумеющееся, но как насчет времени автономной работы, скорости зарядки, поддержки подключения или цены?